CHEM0019 Computational Chemistry: Vibrational Spectroscopy
Hello, dear friend, you can consult us at any time if you have any questions, add WeChat: daixieit
CHEM0019 Computational Chemistry: Vibrational Spectroscopy
The work in this experiment combines the principles of vibrational spectroscopy of diatomic molecules, that you learnt in CHEM0019, with group theory, that you may be learning in elsewhere or can read about in Vincent,“Molecular Symmetry and Group Theory” (which is available as an e-book).
In this experiment, we will use the Gaussian program via the WebMO interface to calculate normal mode vibrations and use these to interpret IR spectra and characterise potential energy surfaces.
Logon to WebMo by going to webmo.chem.ucl.ac.uk in a browser.
1 The IR spectrum of water
1. Start a new job.
2. Draw a water molecule in the build window.
3. Clean up the structure using “Comprehensive - Mechanics”
4. Symmetrise the molecule. Select “Calculate → Symmetry → Symmetrise Molecule” . A window should open with the detected symmetry - it should recognise that water is C2v . Click on “Sym- metrise”. Then “OK” .
Move on to submit a calculation, by pressing the arrow at the bottom right. Call it H2O 631 and select an “Optimise + Vib Freq” Hartree-Fock calculation with a 6-31G(d) basis set. Submit this job.
an “Optimise + Vib Freq” Hartree-Fock calculation with a 6-31G(d) basis set. Submit this job.
Repeat this process to calculate the frequencies at the MP2 (MollerPlesset2) level. Call the calculation H2O MP2 and select a theory level of MP2 .
.
In the results window scroll down to find “Vibrational Modes” is the list of frequencies for the water molecule. View each in turn. The magnifying glass draws arrows to represent the vibrational motion. The film strip animates the vibration - see how the motion corresponds to the arrows. The vibrations are classified as “bend”, “symmetric-stretch” and “anti-symmetric-stretch”. Identify which is which and fill out the following table with the frequencies (harmonic wavenumbers) from both Hartree-Fock (HF) and Moller-Plesset (MP2) calculations. The experimental spectrum has bands at 1595, 3657, and 3756 cm − 1 . Add these values to the table, assigned to the relevant vibration, and comment on how they compare with the calculated values.

For a full report, you should consider if, and if so why, the type of calculation has an impact and whether the error is significant, systematic or irrelevant.
2 The structure of ClF3
The IR spectrum can be used to determine the geometry of a molecule. From bonding considerations, ClF3 could be in one of 2 geometries, either T-shaped or triangular. Using point group notation these are C2v and D3h respectively.
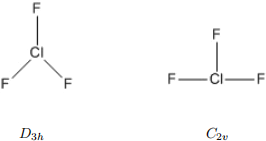
Set up “Optimise + Vib Freq” calculations for the 2 possible structures calling them ClF3 C2V and ClF3 D3H. Use the 6-31G(d) basis set and the MP2 level of theory. To get the correct symmetries:
ClF3 D3H. Use the 6-31G(d) basis set and the MP2 level of theory. To get the correct symmetries:
1. Draw the ClF3 molecule in the build window in the desired shape.
2. Clean up using “Comprehensive - Idealised”. Do not use Mechanics or it will always go to D3h.
3. Symmetrize the molecule. Select “Calculate → Symmetry → Symmetrize Molecule” . A window will open with the detected symmetry - it should be what you want. If not, try again.
Fill out the following table with the 6 harmonic wavenumbers for the 2 structures, including a charac- terisation of each. Use descriptions such as “stretch”, “bend”, “wag”, “rock”, “in-plane”, “out-of-plane” “symmetric”, and “anti-symmetric” .

Below is a table of the measured peaks in the IR and Raman spectra of ClF3. Using the calculated wavenumbers and the IR intensities, determine which of the structures is correct, i.e. which one best reproduces the experimental spectrum.
Structure of ClF3 :
Assign the experimental lines to the vibrational modes.

Finally, Calculate the difference in energy in (i) kJ mol − 1 and (ii) eV between the 2 structures at both the RHF and MP2 levels (1 Hartree = 2625.5 kJ mol − 1 ). Write these values below.
For a full report, you should discuss the comparison between experiment and theory.
3 The NH3 barrier to inversion
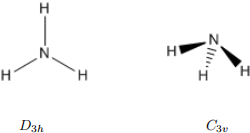
Set up ammonia in a pyramidal (C3v) and a planar (D3h) geometry: NH3 C3V and NH3 D3H. Run
 calculations to do the vibrational analysis for the two structures at the MP2/6-31G(d) level. List and describe the vibrations in the table below.
calculations to do the vibrational analysis for the two structures at the MP2/6-31G(d) level. List and describe the vibrations in the table below.

1. The labels have changed on going from the pyramidal C3v structure to the planar D3h structure, but the vibrational modes have not changed dramatically. Correlate the modes as the structure is changed by listing the vibrations of the C3v structures according to symmetry label and mapping them to the corresponding modes in the D3h structure.

2. Which of the structures is at the minimum energy? Which is at a transition state? How can you tell?
3. The reaction coordinate is the vibration at the transition state with an “imaginary frequency” (listed as a negative wavenumber). This vibration is able to change one structure into the other. Sketch a potential energy surface along the reaction coordinate. It should have 2 minima and a transition state in the middle. Sketch structures to represent ammonia in the three forms (the two minima and the transition state). Add arrows to represent the reaction coordinate vibration. Finally, add the barrier height, i.e. the energy difference between the structures at the MP2 level, calculated in kJ mol − 1 and eV.
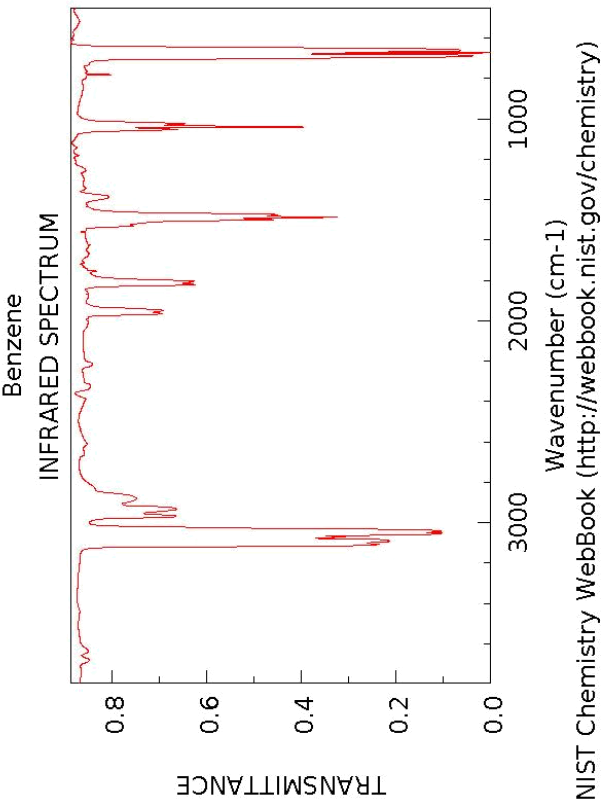
4 The IR spectrum of benzene
The experimental IR spectrum of benzene is reproduced on the next page. Calculate the vibrational modes of benzene at the MP2/6-31G(d) level.
1. With a sketch depicting the vibrational mode, and appropriate description, assign the bands. You should be able to assign four bands. Note that some modes are degenerate - draw only one of the pair.
2. Some of the bands have not been assigned (e.g. those around 2000 cm − 1 . Unassigned bands are usually due to hot bands and combination bands - explain the meanings of these terms.
3. The bands all have a double peak structure. Why? Hint: think about molecular motions other than vibrational.
5 The IR spectrum of aniline (C6H5NH2 )
This exercise is only required to be completed for those selected to write a full report on this experiment.
Predict whether aniline (amino benzene) is planar or pyramidal around the N-atom. You must explain in detail how you arrive at your conclusion; for example, you may like to explain which calculations you performed, on which geometries, and the results of these calculations (energy, optimised geometry, vibrations).
2024-01-23