Bioinformatics, interpretation and data quality assurance in genome analysis: Data Handling Assessment
Hello, dear friend, you can consult us at any time if you have any questions, add WeChat: daixieit
Bioinformatics, interpretation and data quality assurance in genome analysis: Data Handling Assessment
From raw data to alignment and variant calls
The assessment is designed to:
· Test your ability to run standard NGS pipeline using galaxy.
· Test your basic knowledge of a standard NGS pipeline.
· Test your understanding of file formats associated with NGS analysis.
· Test your ability to extract relevant information from fastq, BAM/SAM and VCF files.
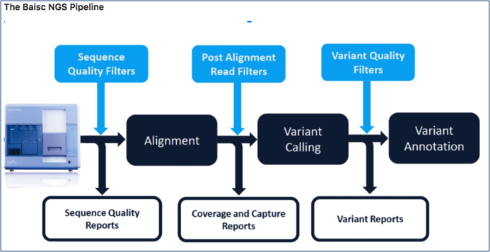
You have been provided with paired end fastq data and an annotation bed file from an Illumina HiSeq 2500 run. Using Galaxy (https://usegalaxy.org/) execute a standard Bioinformatics NGS pipeline to perform read alignment, variant discovery and annotation as described in the following NGS Pipeline section. You are required to share your Galaxy history and published workflow with the examiners by submitting them as web links in this report. Based on the results and output from your pipeline, provide the following information and answer each question. The pipeline is expected to work from end to end without producing errors. If the pipeline includes some manual steps to be performed externally (e.g. using wAnnovar or VEP for variant annotation), the workflow can either stop at that step or take an externally generated sample file (e.g. the wAnnovar output) and run end to end with it. Please note that all steps of the pipeline, including the manual uploads, should be present in at least one submitted Galaxy history. We will test your pipeline by processing a subset of the provided data.
Fastq Read 1 (~750MB): https://s3-eu-west-1.amazonaws.com/workshopdata2017/NGS0001.R1.fastq.qz
Fastq Read 2 (~750MB):https://s3-eu-west-1.amazonaws.com/workshopdata2017/NGS0001.R2.fastq.qz
Annotation File (10M): https://s3-eu-west-1.amazonaws.com/workshopdata2017/annotation.bed
1. The NGS Pipeline (50pts – 45% of final mark)
1.0 Share Your working Galaxy History & Workflow for testing (10pt will be given to a fully working pipeline)
1. Galaxy Shared History (URL):
2. Galaxy Shared Workflow (URL):
Implement and run the following NGS Pipeline:
1.1. Pre-Alignment QC (4 pts)
· Upload raw sequencing reads and annotation data (1pt)
· Perform quality assessment and trimming (2pt)
· Perform basic quality assessment of paired trimmed sequencing data (1pt)
1.2. Alignment (15pts)
· Align the paired trimmed fastq files using bwa mem and reference genome hg19 (3pts)
· Edit your bwa mem step to include read group information in your BAM file (4pts)
· Perform duplicate marking (2pts)
· Quality Filter the duplicate marked BAM file (2pts)
· Generate standard alignment statistics (i.e. flagstats, idxstats, depth of coverage, insert size) (4pts)
1.3. Variant Calling (2pts)
· Call Variants using Freebayes(1pt)
· Quality Filter Variants using your choice of filters (1pt)
1.4. Variant Annotation and Prioritization (4pts)
· Annotate variants using either wANNOVAR or VEP and download the results in your preferred format. Open the file with excel (or any other spreadsheet software) and save – upload this with your assessment. Then upload the wAnnovar/VEP results onto Galaxy in the appropriate format to perform the remaining steps of the pipeline. (Use an academic email address if submitting a wANNOVAR job) (2pts)
· Perform basic variant prioritization: select (filter) rare exonic variants. Please explain your choice of selection criteria (2pts).
1.5. Using an alternative tool for Variant Annotation and Prioritization (10pts)
· Copy your Galaxy history and rename it accordingly (1pts)
· Using the quality-filtered Freebayes variants, i.e. not annotated with wAnnovar or VEP, annotate variants using SnpEFF and/or SnpSift using the Galaxy platform (6pts).
· Perform basic variant prioritization again using the variants annotated with snpEFF and/or SnpSift: filter to rare exonic variants. Please explain your choice of selection criteria (3pts)
· Share your new Galaxy History and Workflow:
o New Galaxy Shared History (URL):
o New Galaxy Shared Workflow (URL):
1.6. Using alternative tools for alignment or variant calling (5 pts)
· Copy you Galaxy history and rename it accordingly (1pts)
· Modify the pipeline by replacing either the aligner or the variant caller with an alternative tool, while leaving the rest of the pipeline unchanged. Run it to make sure that it works end-to-end and share it below (4.pts)
o New Galaxy Shared History (URL):
o New Galaxy Shared Workflow (URL):
2. Bioinformatics Assignment Basic Report (60pts – 55% of final mark)
Please report on and answer the following:
1. Which platform was used for sequencing (1pt)
2. What was the flowcell id? (1pt)
3. What lane of the flow cell was the data run on? (1pt)
4. What is the Min & Max read length pre and post quality trimming? (2pts)
5. How many raw reads are there? (1pt)
6. How many reads are retained in the paired trimmed/filtered? (2pts)
7. What percentage of reads were lost during the quality trimming step (show your working out)? (1pt)
8. What Chromosomes do the reads align to? Provide a table including the chromosome name and the number of reads mapping to each chromosome. Use the template below (extend/edit as needed)? (2pts)
a. What software was used to determine this? (1pt)
Use the table template below (extend/edit as needed)
|
Chromosome |
N Mapped Reads |
|
Chr1 |
|
|
Chr2 |
|
|
… |
|
|
… |
|
9. What is the average insert size in the final filtered .bam file? (1pt)
a. What software was used to determine this?(1pt)
10. Based on your observations, is the data set from i) a Whole Exome panel or ii) a clinical gene panel or iii) a whole genome? (1pt)
a. What evidence supports your answer? (3pt)
11. How many variants are identified by Freebayes pre- and post- variant filtering with VCFfilter? (2pts)
|
N Variants Freebayes (raw) |
|
|
N Variants Freebayes (filtered) |
|
12. What information/fields in the VCF file did you use to perform the variant filtering and why? Report the filtering expression you used (2pts)
13. Provide a table reporting on the number of SNPs (SNVs) and INDELS (both insertions and deletions), detected by Freebayes (2pts). Use the template below.
|
SNPS |
|
|
INDELS |
|
14. For every major step of the NGS pipeline provide a table reporting:
a. Name of the NGS software/tool
b. The software/tool version
c. Assign each tool to one of the following categories: quality control, aligner, variant caller, variant annotator or general.
d. Indicate the format of the input and output of each tool (FASTQ, SAM/BAM, VCF or HTML/REPORT)
Use the table template below (extend/edit as needed – I have started this for you as an example) (5pts)
|
Tool |
Version |
Input |
Output |
Category |
|
Fastqc |
????? |
FASTQ |
HTML/REPORT |
quality control |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
15. What file format are the raw NGS reads provided as? What file format are the aligned reads saved/stored as? What file format are the variant calls stored as? (1pt)
16. Why is variant filtering important? (2 pts)
17. Why is variant prioritization important? (1 pts)
18. From the wANNOVAR, VEP or snpEFF output, how many variants are exonic? (1pt)
19. From the wANNOVAR, VEP or snpEFF output, how many variants are reported as stop gain? (1pt)
20. From the wANNOVAR, VEP or snpEFF output, how many variants are reported as nonsynonymous SNV? (1pt)
21. Use your variant annotations to identify rare nonsynonymous SNVs (Please describe/list your filtering terms and steps). List them and their annotated gene below and report the standard HGVS annotation for the canonical transcript along with the basic gene description and report the Variant call quality, genotype, and genotype depth. (4pts)
Use the table template below (extend/edit as needed)
|
Gene |
Description |
QUAL |
Genotype & (GT:DP) |
HGVS |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
22. If more that one gene has been identified, pick one, and briefly identify any one or mores diseases this gene may have been associated with. Provide the OMIM ID and at least one reference for the reported association. Determine if any of the reported variants could contribute to this disease. Provide a brief explanation for your conclusion with references where appropriate. (4pts)
23. View the BAM file in either IGV or ucsc genome browser. Focus on one of the genes selected in question 24 and paste a screen shot or saved image below (2pts).
24. Why would we want to visualize the read alignments in this way - what kind of information can we gain? (3pts)
25. In the previous section of the assignment (The NGS Pipeline), you have been asked to run the pipeline replacing either the aligner or the variant caller with an alternative tool, while leaving the rest of the pipeline unchanged. As a result, you should have two sets of variant calls, one from the original pipeline and one from the “modified” pipeline. Using these two sets, please answer the following questions:
a. What is the number of variants in the union of the two sets before annotation (after quality filtering)? (3pts)
b. What is the number of variants in the intersection of the two sets after annotation and prioritization (i.e. after filtering for variants that are exonic and rare)? (4pts)
c. With respect to question 22 of this section, how different (if at all) would your answer to question 22 be if you used the alternative tool? (4pts)
2024-01-09